
Budding yeast is one of a widely used model organism for aging study. The cells of budding yeast asymmetrically divide to produce daughter cells from mother cells. The finite number of the daughter cells are generated from a mother cell. Mother cells no longer divide after production of 20-30 daughter cells. The number of division cycles is defined as replicative lifespan (Fig 1).
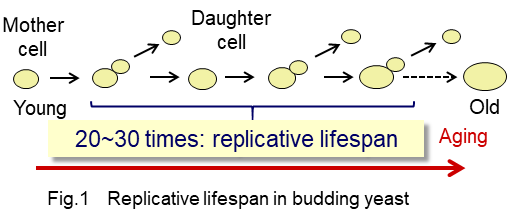
In contrast, newly born daughter cells are rejuvenated and then can divide from 20 to 30 times. Asymmetry of cellular components between mother and daughter cells is known to be involved in replicative lifespan. Dysfunctional organelles, aggregated proteins, and extrachromosomal ribosomal DNA circles are accumulated in mother cells in progress of cell division. However, it has been poorly understood what kind of differences in quality and quantity of individual proteins are accumulated and how such asymmetry can affect aging processes.
In order to reveal relevant proteins associated to asymmetry aging, we have constructed a method in which qualitative difference in proteome between mother and daughter cells can be analyzed (Fig. 2) (Okada et al. 2017).
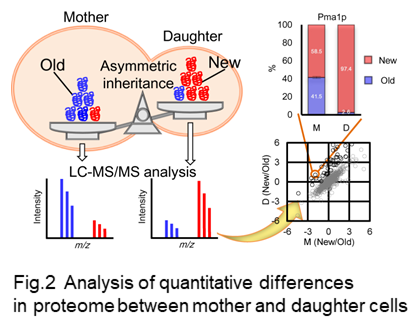
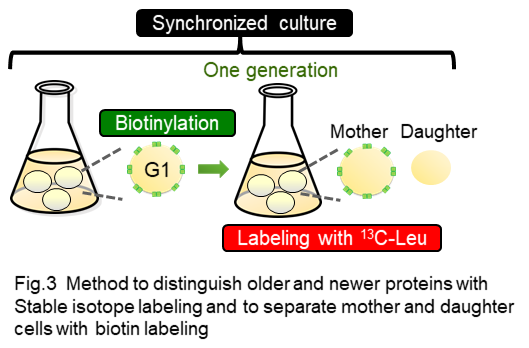
In this method, newly synthesized proteins are labeled with stable isotope amino acid during synchronized culture to allow us to distinguish older and newer proteins with mass spectrometric analysis (Fig 2, Fig 3). Biotin is used for labeling of mother cells to separate mother and daughter cells after one round of cell cycle is completed (Fig 3).
We successfully identified about 20 proteins that are older in mother cells as compared with daughter cells (Fig 4) (Okada et al. 2017).
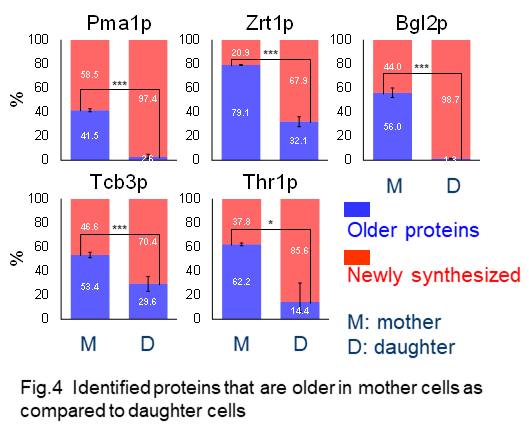
The involvement of identified proteins in aging process remains to be elucidated by further studies, proteomic analysis in short or long-lived mutant and rejuvenation of older proteins.
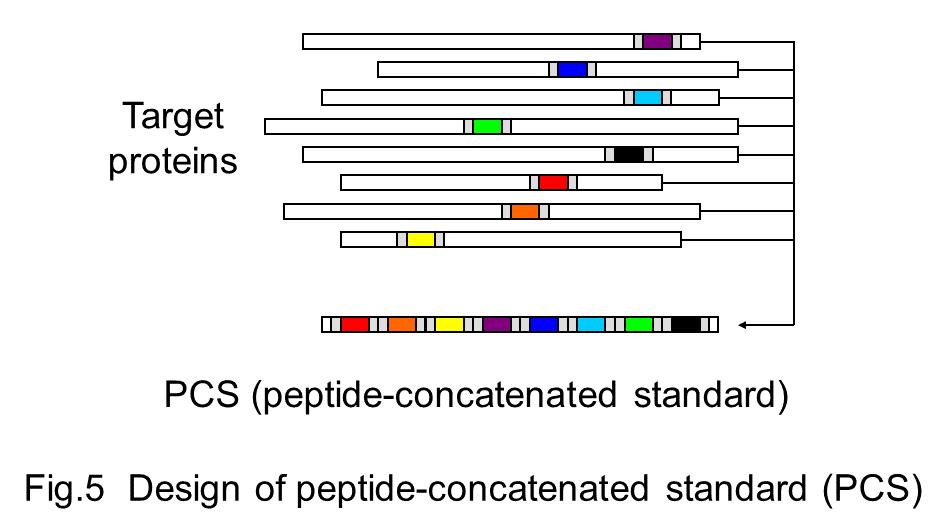
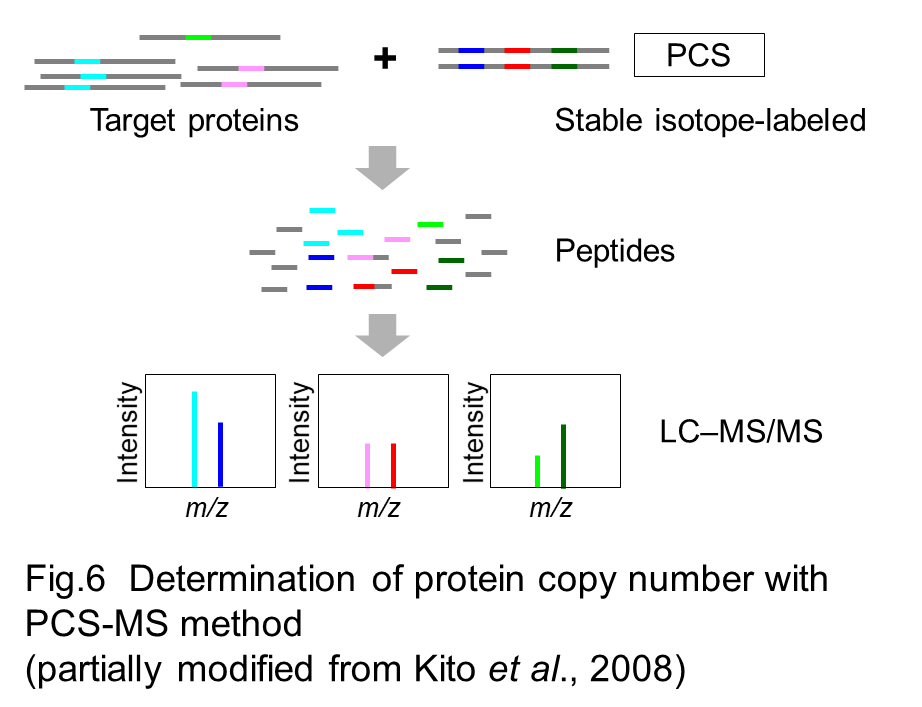
Comprehensive analysis of protein copy number per cell with high accuracy is critical to understand proteome and its relevance to a variety of biological processes. Many kinds of strategies and techniques based on mass spectrometry have contributed to such kind of quantitative proteomics analysis. We have developed an original strategy, PCS-MS method, in which peptide-concatenated standard (PCS) is used as a stable isotope-labeled internal standard to know the abundance of proteins of interest (Kito et al. 2007, Kito et al. 2016). Peptides suitable for detection and quantification with mass spectrometry are selected from target proteins, and then concatenated into a single artificial protein to construct PCS (Fig 5). Protein sample is mixed with known amount of stable isotope-labeled PCS to measure the copy number of proteins from the ratio of peptide intensities between target proteins and PCS (Fig 6).
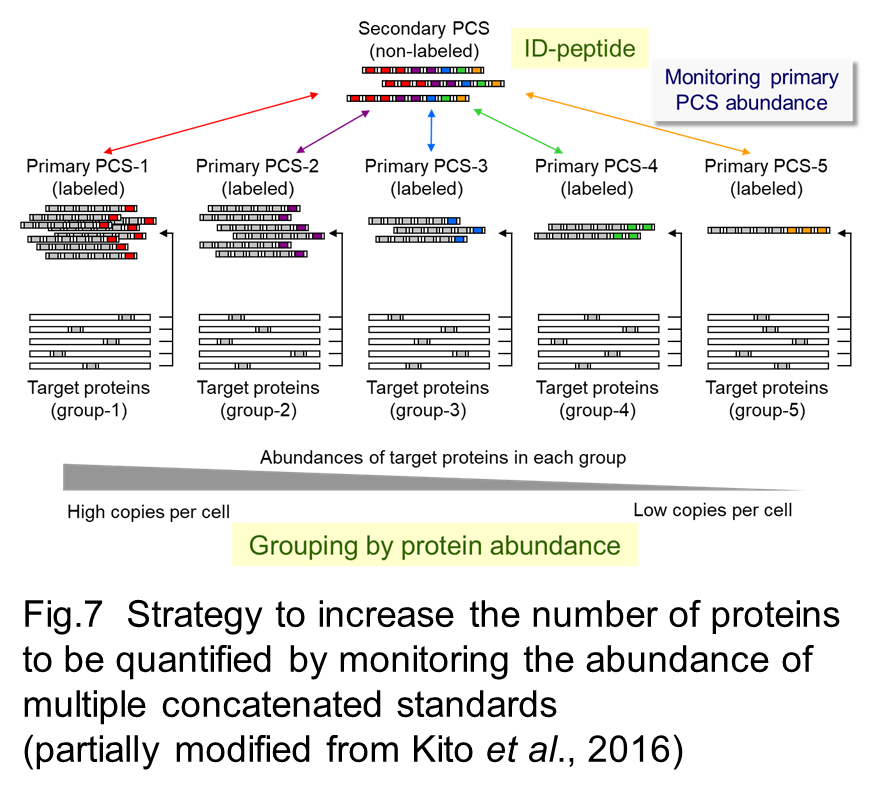
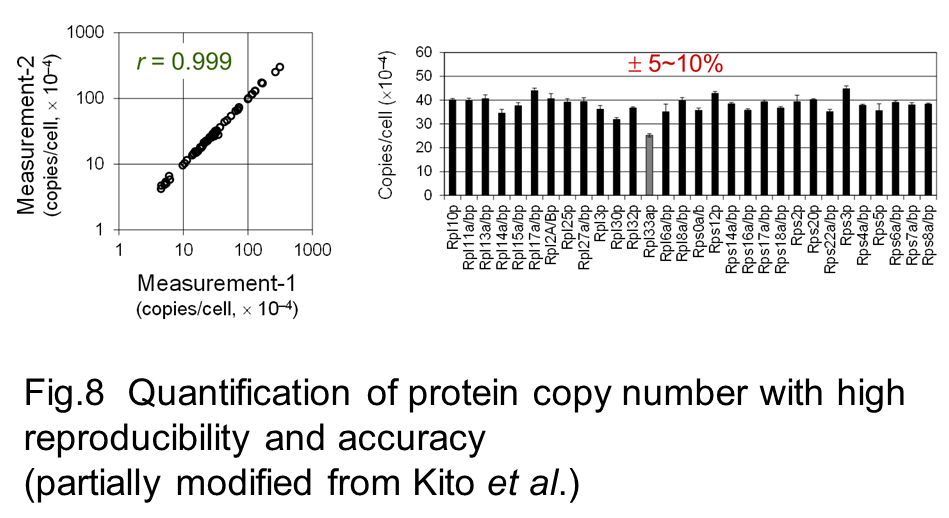
We have demonstrated that this strategy allows accurate quantification of subunit stoichiometry in protein complexes and their dynamic change in progress of cellular processes (Kito et al. 2007, Kito et al. 2008). Furthermore, integration of a system to monitor the exact amounts of multiple PCSs successfully achieved the increase in the number of proteins to be quantified and expansion of the dynamic range (Fig 7, Fig 8) (Kito et al. 2016).
We are now progress with the development of workflow that allows the quantification of proteins that are intrinsically difficult to detect.
A verity of yeast species within the Saccharomycetaceae family are an interesting group of model organism to study the impact of global differences in metabolic processes in response to change in nutrient conditions and understand the evolutionary role for duplicated genes known to be involved in the species-specific evolution in variable groups of organisms. Saccharomyces cerevisiae and very closely related yeast species can efficiently produce ATP through glycolytic process regardless of presence or absence of oxygen (Fig 9). In contrast, yeast species in the other genus have high metabolic activity of TCA cycle and oxidative phosphorylation in mitochondria to generate ATP (Fig 9). Yeast species in the Saccharomyces genus have about 500 pairs of duplicated genes that arose through whole genome duplication, thus chromosome of this group of yeast is about twice as number as that of other groups (Fig 9).
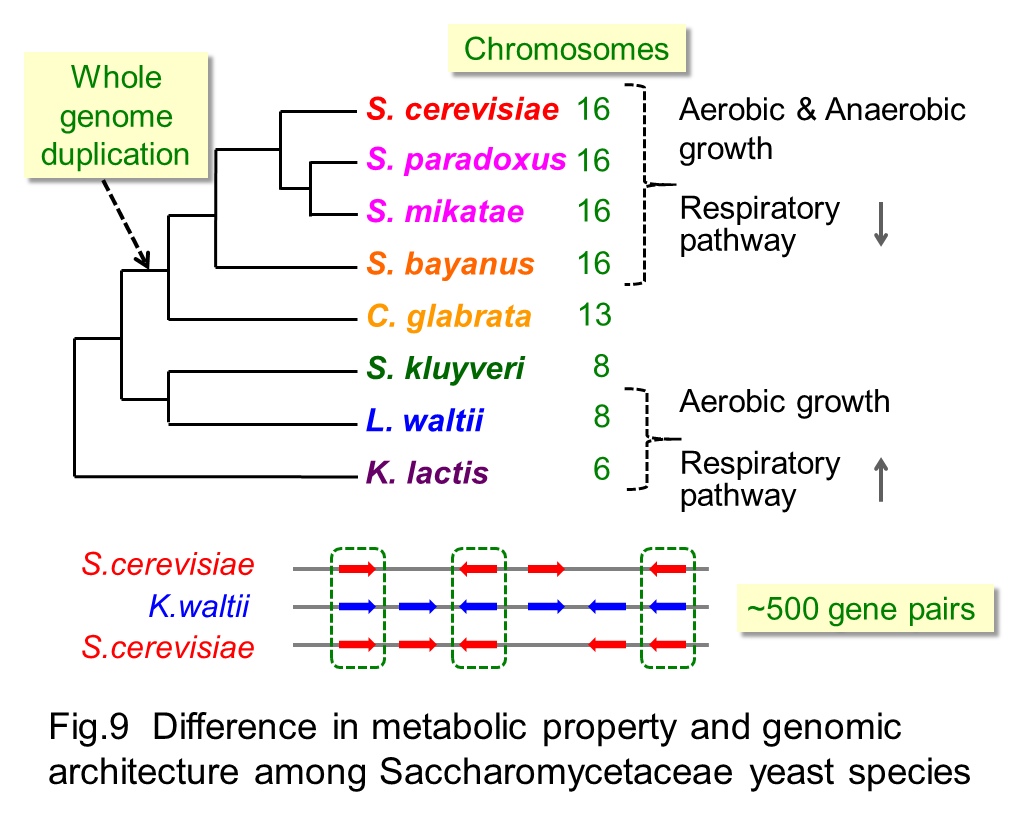
In our laboratory, comparative proteomic analyses across these yeast species (Fig 10) with distinct metabolic propensity and genomic architecture revealed differences in abundance of metabolic enzymes (Fig 11) and conserved abundance between duplicated and non-duplicated paralogues (Kito et al. 2016). It is also interesting that yeast species with higher growth rate have lower abundance of the metabolic enzymes that is not required for cell growth and higher abundance of protein groups involved in the protein synthesis.
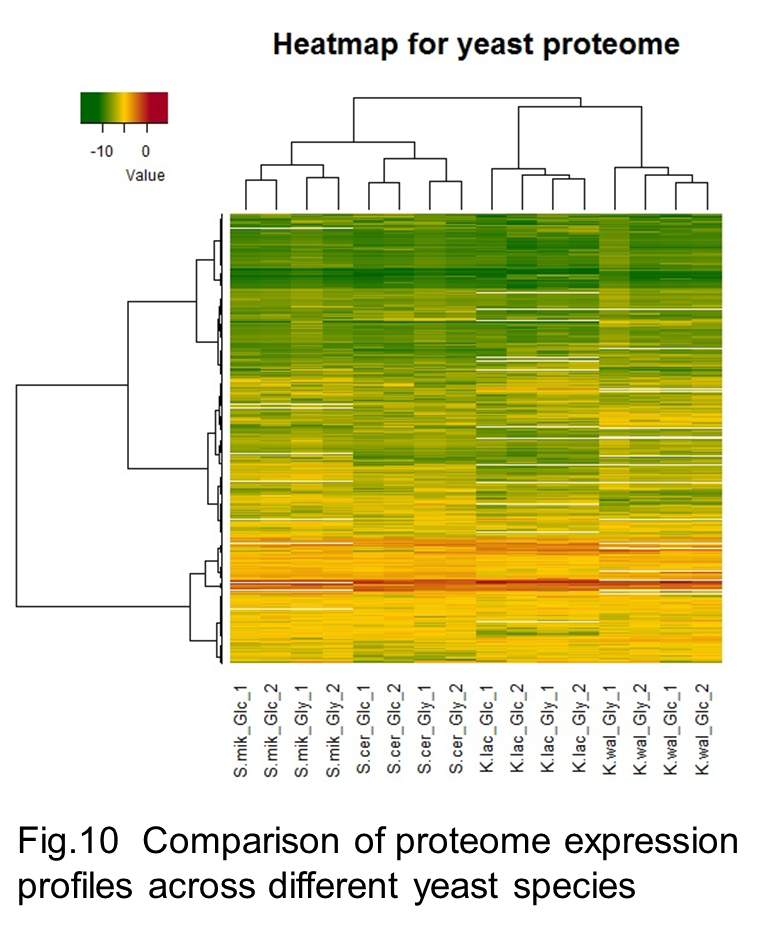
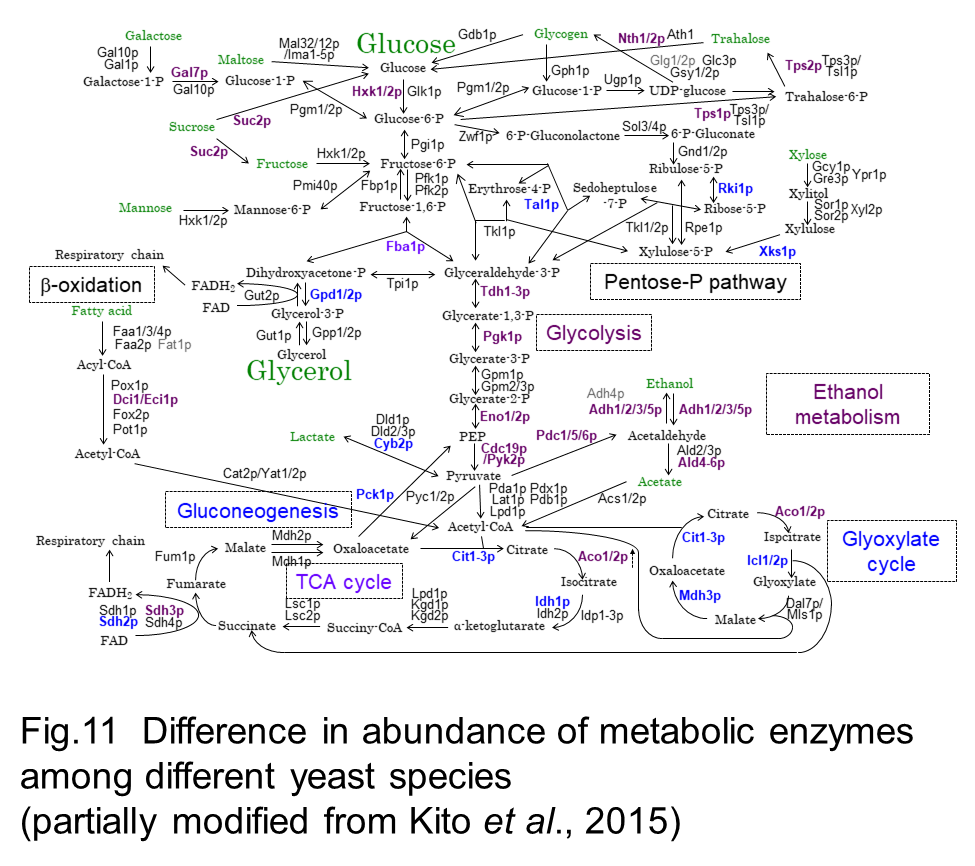
We are now in the study to reveal the significant of proteome balance between catabolic and anabolic processes and are also expanding inter-species comparative proteomics across more yeast species.